Неврология — Нервные болезни
Метахроматическая лейкодистрофия — аутосомно-рецессивно наследуемая демиелинизирующая патология ЦНС, отличительной особенностью которой является метахроматическое окрашивание зон демиелинизации. В клинике преобладает задержка развития, парезы, судорожные приступы, экстрапирамидные и мозжечковые расстройства, нарушение зрения. В ходе диагностики проводится исследование цереброспинальной жидкости, уровня сульфатидов, активности арилсульфатазы А, КТ/МРТ головного мозга, генетические обследования. Возможна пренатальная диагностика. Лечение симптоматическое, в отдельных случаях проводится трансплантация пуповинной крови или стволовых клеток.
Метахроматическая лейкодистрофия (МЛД) является типичным заболеванием с поражением миелина в виде демиелинизации. МЛД — собирательное название гетерогенной группы фатальных патологий, характеризующихся скоплением сульфатидов в центральной и периферической нервной системе, что приводит к диффузной демиелинизации.
Скопление метахроматическо-реагирующей липидной субстанции происходит в глиальных клетках, макрофагах, нейронах бледного слоя, зубчатого ядра мозжечка, ядрах черепных нервов, а также клетках внутренних органов (почек, желчного пузыря (ЖП), поджелудочной железы (ПЖ), надпочечников, печени).
Метахроматическая лейкодистрофия вызывается дефицитом фермента арилсульфатазы А и относится к группе сфигнолипидозов — лизосомных болезней накопления, которые характеризуются скоплением в тканях органов-мишеней сфигнолипидов. Другие известные названия заболевания: сульфатидный липидоз, синдром Гринфилда, болезнь Шольца, болезнь Геннеберга, синдром Шольца — Бильшовского — Геннеберга. Одно из своих названий — болезнь Шольца — МЛД получила в честь немецкого невропатолога и психиатра Вилибальд Оскара Шольца, который в 1925 описал семейный случай ювенильной формы этого заболевания. В 1928 г. немецкие неврологи Макс Бильшовский (1869-1940) и Ричард Геннеберг (1868-1962) описали то же заболевание и впервые предложили термин «лейкодистрофия» для нейродегенеративных заболеваний белого вещества. Также в 1928 эти исследователи предложили первую классификацию лейкодистрофий, основанную на клинических, патоморфологических и гистохимических критериях. Поэтому по гистохимическим критериям все лейкодистрофии были разделены на ортохроматические и метахроматические. Термин «ортохроматические» применяется для описания образцов ткани, которые окрашиваются в тот же цвет, что и используемый для этого краситель, «метахроматические» означает окраску гистологических структур в цвет, несвойственный этому красителю. В честь этих ученых ювенильная метахроматическая лейкодистрофия получила одно из своих названий — «болезнь Шольца — Бильшовского — Геннеберга». Макс Бильшовский также является соавтором описания поздней инфантильной формы нейронального цероидного липофусциноза, известного как болезнь Бильшовского — Янского.
В то же время эпоним «болезнь Шольца» почти не используется, что, по мнению некоторых специалистов, связано с некоторыми фактами биографии этого медика. Вилибальд Шольц (1889-1971) получил степень доктора 1914 года в Йене, позже возглавил клинику нервных и психических болезней в г. Лейпциг. С 1935 по 1961 гг. он возглавлял Научно-исследовательский институт психиатрии в г. Мюнхен. Во время Второй мировой войны В. Шольц опубликовал не менее 11 работ на основе исследования мозга 194 человек — жертв нацистских программ эвтаназии «Т-4», которая осуществлялась в вышеупомянутом мюнхенском Научно-исследовательском институте психиатрии. Проект умерщвления «Т-4» («Акция Тиргартенштрассе 4») — наименование евгенической нацистской программы по стерилизации, а затем и физуничтожения душевнобольных, умственно отсталых и генетически несовершенных лиц. В институте г. Мюнхен его коллегой был известный врач Юлиус Галлеворден, который, используя медпрепараты головного мозга жертв программы «Т-4», изучал причины атрофии мозжечка, рассеянного склероза и хореи Гентингтона. Известно, что Галлеворден лично присутствовал при уничтожении некоторых жертв и самостоятельно изымал мозг после «эвтаназии». Зато Шольц интересовался преимущественно поисками этиологии болезней, психозов и сенильной деменции. В 1956 Шольц стал главным редактором и автором неврологического раздела фундаметального «Руководства по патологической анатомии и гистологии», куда вошли описания некоторых его наблюдений, сделанные во времена нацизма.
Читать также Миопатии — причины, симптомы, лечение, диагностика, формы
В 1964 американский невролог Джеймс Остин с соавторами установил наличие при МЛД дефекта лизосомального фермента арилсульфатазы А, который приводит к накоплению кислых липидов в тканях пациентов и приводит эффект метахромазии.
Общераспространенность метахроматической лейкодистрофии составляет 1:40 000 — 1: 100 000 человек. Значительно выше заболеваемость в некоторых изолированных популяциях, в частности у евреев Хаббана, популяции евреев, иммигрировавших из Йемена в Израиль, распространенность же составляет около 1,3%. Распространенность поздней инфантильной формы оценивается в 1: 400000 — 1: 170000 новорожденных.
Причиной МЛД является дефицит фермента арилсульфатазы А, который отвечает за гидролиз сульфатидов, от цереброзидов-3-сульфат или 3-О-сульфогалактозилцерамида до галактоцереброзида и сульфата. Всего известно 3 типа арилсульфатазы — А, В (лизосомальные) и С (микросомальные). Патологическое накопление сульфатидов в нервной системе (миелине, нейронах и глии) приводит к поражению белого вещества центральной и периферической нервной систем, что вызывает прогрессирующую деменцию, неврологические отклонения и слепоту. Метахроматические гранулы, которые накапливаются в ЦНС, являются высокотоксичными, приводят к дегенерации миелина и потери аксонов.
Диффузная демиелинизация происходит преимущественно в полушариях большого мозга, при этом серое вещество не поражается. Также происходит сегментарная демиелинизация периферических нервов и накопление метахроматических гранул в шванновских клетках, что вызывает сенсомоторную полиневропатию, нарушение походки и атаксию. При метахроматической лейкодистрофии формируется диффузная симметричная демиелинизация полушарий большого мозга и мозжечка при аномалии олигодендроглии, что приводит к аксональному разрушению, дегенерации пирамидных путей с изоморфным глиозом и демиелинизирующей полинейропатией. Сохраненными остаются только субкортикальные U-волокна. Также метахроматические гранулы, содержащие сульфатиды, накапливаются в других тканях организма, в частности желчевыводящих протоках печени, ЖП, печени, ПЖ, яичниках, лимфатических узлах, глазах, зубной пульпе и дистальных канальцах почек. Повышенный уровень этих соединений также обнаруживается в моче.
Дефектный ген при метахроматической лейкодистрофии расположен на длинном плече 22-й хромосомы (22q13.31-qter) или при нехватке белка-активатора сфигнолипидов — на коротком плече 10-й хромосомы (10q21- q22); тип унаследования — аутосомно-рецессивный.
Клинически картина метахроматической лейкодистрофии может быть существенно вариабельной у разных детей, что связано со значительным полиморфизмом гена ARSA. Всего в группу метахроматических лейкодистрофий входят 5 аллельных форм.
- Поздняя инфантильная (синдром Гринфилда) — наиболее распространенная и тяжелая форма, которая отмечается в 50-60% случаев и дебютирует в 1-2 года (чаще в возрасте 12-18 мес), реже — в 2-4 года. После периода нормального развития начинается регресс моторных навыков и речи. Ребенок становится раздражительным, плохо ест, нарушается походка, появляется и нарастает гипотония мышц, отмечаются частые падения, нечеткость речи, потеря рефлексов (на ранних стадиях), косоглазие, нистагм, мозжечковые нарушения, атрофия зрительных нервов. Первые проявления заболевания могут появляться после перенесенной инфекции или применения наркоза, после чего исчезают на несколько недель и затем снова возвращаются. В большинстве случаев характерно быстрое прогрессирование заболевания с присоединением таких неврологических осложнений, как спастичность (в соединении с признаками разрушения периферических нервов), потеря способности ходить и стоять, эпилептические припадки (миоклонические, парциальные или генерализованные), корковая слепота, падение интеллекта и слуха, псевдобульбарный синдром на фоне децеребрационной ригидности. Затем наступает смерть (обычно через 2-4 года после дебюта).
- Ювенильная форма (болезнь Шольца) возникает в 20-30% случаев. Дебютирует в возрасте 4-16 лет, стартуя с нарушений походки, эмоциональных и поведенческих аномалий. Проявления полинейропатии менее явны, однако отмечается проявление прогрессирующей глухоты и слепоты, афазии, впоследствии формируются и нарастают признаки слабоумия, недержания мочи и кала, спастичность мышц, атаксия, непроизвольные движения; порой формируются судорожные припадки; средний срок жизни составляет 7 лет, большинство пациентов умирают в течение первых 10 лет жизни, однако известны случаи, когда продолжительность жизни таких детей составляла 20 лет и больше.
- Форма взрослых (синдром ван Богарта — Ниссена — Пфайффера) встречается редко. Дебютирует в возрасте 16-60 лет. Характеризуется медленно прогрессирующей деменцией, психозами, атрофией зрительных нервов, атаксией, пирамидно-экстрапирамидной симптоматикой, полинейропатией. В литературе имеются сообщения о случаях, которые проявляются исключительно полинейропатией.
- Частичная недостаточность цереброзидсульфатазы.
- Псевдонедостаточность арилсульфатазы А — синдром, характеризующийся выраженным падением активности арилсульфатазы А при отсутствии клинических проявлений.
Читать также Ишемический инсульт — причины, симптомы, лечение
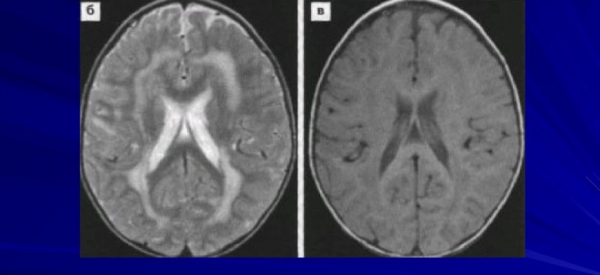
Метахроматическая лейкодистрофия может быть заподозренной у детей с прогрессирующими неврологическими нарушениями и с признаками дегенерации белого вещества по данным МРТ. В типичных случаях на МРТ оказываются диффузные симметричные гиперинтенсивные очаги преимущественно в теменно-затылочных участках белого вещества, которые впоследствии распространяются в направлении лобных участков, комиссуральных волокон мозолистого тела и перивентрикулярного белого вещества. Мерой прогрессирования заболевания являются аномалии на МРТ, которые становятся все более выраженными при рострально- каудальной прогрессии; развивается атрофия мозга. Поражение передних отделов мозга преимущественно характерно для пациентов с поздним началом заболевания. Гипоинтенсивные радиальные полоски сигнала между гиперинтенсивными участками белого вещества формируют МР-симптом «тигровой кожи» на Т2-взвешенных аксиальных срезах. Сохраненные периваскулярные участки белого вещества перивентрикулярных зон и полуовального центра формируют гипоинтенсивные пятна, напоминающие шкуру леопарда.
Важным для установления диагноза является обнаружение снижения активности ARSA менее чем на 10% от нормы в лейкоцитах крови или культуре фибробластов кожи. Однако при использовании только оценки активности ARSA возможные ошибки в диагностике, так как бывают пациенты с псевдодефицитом этого фермента. При таких состояниях активность ARSA снижена на уровне 5-20% от нормы, однако клинические проявления метахроматической лейкодистрофии отсутствуют. Также уровень активности ARSA не может быть использован как предиктор тяжести МЛД, так как не всегда дефицит фермента выражен и сопровождается тяжелыми клиническими проявлениями.
Окончательно диагноз метахроматической лейкодистрофии подтверждается с помощью молекулярно-генетического тестирования, которое обнаруживает мутацию гена, кодирующего ARSA. Также возможна верификация диагноза путем выявления экскреции сульфатидов с мочой методами шаровой хроматографии или масс-спектроскопии, возможно и обнаружение метахроматических липидных гранул в биоптатах нервной ткани, что является патогномоничным для метахроматической лейкодистрофии.
Сегодня метахроматическая лейкодистрофия считается неизлечимым заболеванием с фатальным течением. Существует несколько подходов к терапии, преимущественно еще на стадии разработки, в частности трансплантация костного мозга, введение рекомбинантного фермента ARSA (препарат для заместительной терапии «Метазим») или мезенхимальных стволовых клеток, применение варфарина, а также генная терапия.
Согласно МКБ 10, болезни присвоен код Е75.2.
- I стадия
- II стадия
- III стадии
- IV стадии
- Врожденная метахроматическая лейкодистрофия
- Позднедетская метахроматическая лейкодистрофия
- Ювенильная метахроматическая лейкодистрофия
- Взрослая метахроматическая лейкодистрофия
- Диагностика и симптомы болезни в Израиле
- Как лечат: методы лечение метахроматической лейкодистрофии
- Пересадка костного мозга (ТКМ)
- Терапия
- Прогноз после лечения и последствия
I стадия
Первая стадия синдрома метахроматической лейкодистрофии характеризуется слабостью скелетных мышц. У пациента снижается тонус рук и ног, глубокие сухожильные рефлексы, возникает переразгибание коленных суставов и затруднения при ходьбе. Длительность стадии — от нескольких месяцев до года и больше.
II стадия
Вторая стадия характеризуется отставанием в интеллектуальном развитии, нарушением речи, нистагмом (спонтанным движением глазных яблок). Появляется гипертонус нижних конечностей и интермитирующие боли. Ребенок утрачивает возможность стоять. При осмотре глазного дна выявляются признаки атрофии глазного нерва, желтое пятно приобретает серый оттенок. Длительность стадии — несколько месяцев.
III стадии
Третья стадия характеризуется:
- тетраплегией (паралич всех конечностей);
- мышечной дистрофией (постепенная атрофия скелетных мышц);
- декортикацией (функциональное отключение коры больших полушарий);
- децеребрацией (нарушение связей между головным и спинным мозгом);
- бульбарным и псевдобульбарным параличом (расстройства глотания, жевания, мимики, речи и других функций);
- тяжелыми психическими дефектами.
Состояние ребенка постепенно ухудшается.
IV стадии
На четвертой, последней стадии синдрома метахроматической лейкодистрофии исчезает полностью реакция на окружающий мир. Ребенок теряет зрение, перестает говорить, не может жевать и глотать еду, из-за чего кормление возможно только через зонд или гастростому. Продолжительность стадии — от нескольких месяцев до нескольких лет, в зависимости от качества ухода за больным.
Врожденная метахроматическая лейкодистрофия
Как правило, врожденный синдром метахроматической лейкодистрофии манифестирует в трехмесячном возрасте, когда ребенок начинает отставать в психомоторном развитии. Также появляются эпилептические приступы. Болезнь завершается летальным исходом на первом году жизни.
Читать также Спинальная амиотрофия Верднига-Гоффмана — причины, симптомы, диагностика, лечение
Позднедетская метахроматическая лейкодистрофия
Позднедетская, или поздняя инфантильная форма заболевания, проявляется на втором году жизни. У ребенка ухудшается моторика, появляются задержки психического развития в сравнение со сверстниками. Родители замечают нарушения походки, обусловленные снижением тонуса скелетных мышц. Болезнь постепенно прогрессирует и приводит к полной утере реакции на окружающие события, слепоте, невозможности самостоятельно есть. При качественном уходе длительность жизни составляет один – два года.
Ювенильная метахроматическая лейкодистрофия
Данная форма синдрома метахроматической лейкодистрофии дебютирует у детей 3 – 10 лет, чаще всего в шестилетнем возрасте. Дети становятся эмоционально лабильными. Родители отмечают изменения в поведении, снижение когнитивных способностей. У школьников резко падает успеваемость. Появляются атактические расстройства, затрудняется ходьба, отмечаются эпилептические приступы. Летальный исход наступает в возрасте 10 – 15 лет, в зависимости от начала симптоматики.
Взрослая метахроматическая лейкодистрофия
Синдром взрослой метахроматической лейкодистрофии проявляется после периода полового созревания и до 60 лет. Как правило, начинается с психических расстройств — шизофреноподобного или психопатического характера, из-за чего большинство пациентов сначала обращаются за помощью к психиатрам. Гораздо реже патология начинается полиневропатическими расстройствами.
Болезнь прогрессирует медленнее, чем при других формах. Длительность жизни больных — 10 – 20 лет от начала заболевания.
Диагностика и симптомы болезни в Израиле
При диагностике синдрома метахроматической лейкодистрофии крайне важно первичное обследование. Оно включает в себя тщательное изучение истории болезни как маленького пациента, так и его родителей. Врач детально выслушивает жалобы, устанавливает время их появления и динамику, собирает семейный анамнез для установки пути наследования заболевания.
Следующий этап — объективный осмотр. Врач оценивает тонус мышц, рефлексы, походку и координацию движений. Пациента обязательно консультирует ЛОР и окулист. Это необходимо для определения нарушений зрения и слуха.
Из лабораторных и инструментальных исследований при подозрении на синдром метахроматической лейкодистрофии применяют:
- анализ спинномозговой жидкости;
- биохимический скрининг крови;
- нейросонографию;
- эхо-энцефалографию;
- КТ;
- МРТ;
- электромиографию;
Кроме того, на сегодняшний день применяются методы ДНК-диагностики, которые позволяют диагностировать заболевание в период внутриутробного развития плода. В таких случаях семейную пару консультирует врач-генетик.
Как лечат: методы лечение метахроматической лейкодистрофии
К сожалению, эффективных оперативных или терапевтических мероприятий, которые могли бы полностью избавить пациента от недуга, на сегодняшний день не существует. На первый план при синдроме метахроматической лейкодистрофии выходит симптоматическое лечение.
Пересадка костного мозга (ТКМ)
Пересадка костного мозга или стволовых клеток показана при легких формах заболевания, которые характеризуются умеренной симптоматикой. Донором для пациента выступает тщательно обследованный родственник, не имеющий синдрома метахроматической лейкодистрофии. Однако, эти методики только разрабатываются и находятся на стадии клинических испытаний. Только после их завершения можно будет сделать выводы о возможности замедления прогрессирования болезни или полного ее остановки.
Терапия
На первый план при лечении синдрома метахроматической лейкодистрофии у детей в Израиле на первый план выходит симптоматическое лечение и качественный уход. Пациенту назначают обезболивающие средства, парэнтеральное питание, ухаживающих за больным людей обучают профилактике пролежней и дают всесторонние рекомендации.
Ученые Израиля и всего мира разрабатывают новые методы лечения синдрома метахроматической лейкодистрофии у детей, включающие генную, фермент-заместительную и субстрат-снижающую терапию. Кроме того, предпринимаются попытки повысить активность собственного фермента.
Прогноз после лечения и последствия
Так как лечение синдрома метахроматической лейкодистрофии у детей отсутствует, прогноз болезни крайне неблагоприятен. Средняя длительность жизни больных детей — от года до 6 лет, что зависит от формы заболевания. Среди взрослых показатель несколько больше — 10 – 20 лет. Единственный метод профилактики заболевания — тщательное планирование семьи, своевременное обследование пар из группы риска.